Haemoptysis and an unusual ‘lung mass’
Tom Foster 1, Michelle C Williams 2, Peter Maclean 1
- Department of Radiology, Western General Hospital, Edinburgh
- University of Edinburgh, Edinburgh
A young male patient presented to his GP after an episode of haemoptysis. He reported a 1 month history of dyspnoea, non-productive cough and intermittent fever. Initial chest X-ray (Figure 1) showed a 3.5 cm diameter rounded opacity in the right lower zone, with further patchy opacification in the left lower zone. This was initially reported as likely infective consolidation, but the clinical team’s differential diagnosis also included pulmonary embolus or malignancy.
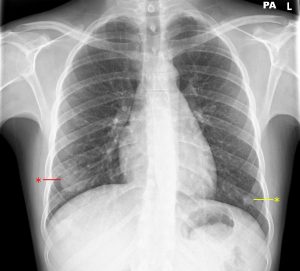
Figure 1. 3.5 cm rounded opacity in the right lower zone (red asterisk), with further patchy opacification in the left lower zone (yellow asterisk).
Further questioning identified a history of weight loss and night sweats. Therefore a CT chest / abdomen / pelvis was performed.
Arterial phase imaging through the lower lungs (Figure 2) showed a 3.5 cm contrast-filled lesion in keeping with a pulmonary artery aneurysm arising from a segmental branch of the right lower lobe pulmonary artery. Lung windows show some surrounding ground-glass change (Figure 3). Coronal MIP imaging (Figure 4) demonstrates a right lower lobe segmental artery pulmonary aneurysm clearly, with contrast within the lesion seen in continuity with the pulmonary trunk. A pulmonary vein is seen adjacent to the lesion, draining to the right atrium. There were a number of other areas of patchy areas of opacification located peripherally in the lungs (Figure 5). Small areas of pulmonary thrombus were seen proximal to some of these lesions. These were therefore felt to represent a mixed picture of multiple small areas of pulmonary haemorrhage, sub-segmental pulmonary emboli and focal areas of pulmonary infarction.
CT imaging of the abdomen (Figure 6) showed multiple wedge-shaped low-attenuation areas throughout both kidneys in keeping with perfusion abnormalities. There was however no other evidence of aneurysm or other significant findings in the abdomen or pelvis.
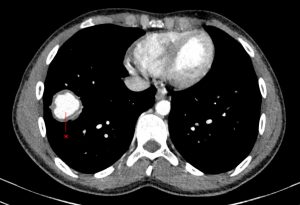
Figure 2. Axial CT slice through a contrast-filled lesion (red asterisk) in the right lower lobe on arterial-phase imaging.
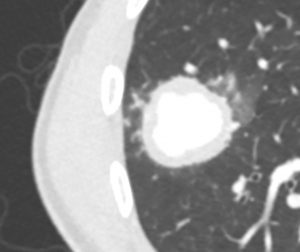
Figure 3. Cropped and magnified view of the lesion on lung window showing surrounding ground-glass opacification.
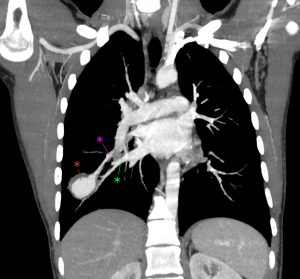
Figure 4. Coronal MIP view of the aneurysm (red asterisk) of a right lower lobe segmental branch artery. The artery proximal to the aneurysm is seen superiorly (pink asterisk) and a draining pulmonary vein is seen inferiorly (green asterisk).
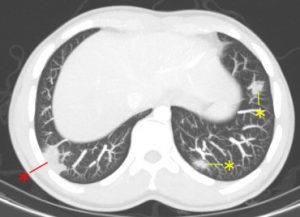
Figure 5. There were multiple other pulmonary abnormalities (asterisks), all located peripherally within the lungs. Some of these (red asterisk) had pulmonary thrombus proximal to them and were felt to represent pulmonary infarction.
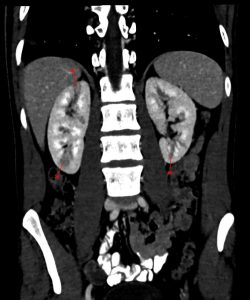
Figure 6. An oblique coronal view through the kidneys, with multiple wedge shaped low attenuation areas bilaterally (arrowed).
The impression at this stage was that findings may represent an underlying vasculitic process such as granulomatosis with polyangiitis (previously called Wegener’s granulomatosis), Takayasu’s arteritis or Behçet’s disease. After discussion with interventional radiology and cardiothoracic surgery, the patient underwent right lower lobe basal segmentectomy.
Histopathology of the resected lung demonstrated a necrotising inflammatory process associated with areas of pulmonary infarction and focal lymphocytic vasculitis of branches of the pulmonary artery. Findings were in keeping with either Behçet’s disease or Hughes-Stovin syndrome (a rare auto-immune condition similar to Behçet’s characterised by thrombophlebitis and pulmonary/bronchial aneurysms). The patient reported oral ulcers, but no other classical findings of Behçet’s disease, therefore the final diagnosis was felt to be either atypical Behçet’s disease or Hughes-Stovin syndrome.
Discussion
Behçet’s disease is a multi-system autoimmune disease of unknown aetiology. It is more common in males and highest incidence in individuals aged 20-30, particularly those in the Middle East and Japan 1. Behçet’s Disease is likely the result of a complex interplay between various genetic and environmental factors (including certain infections and their antigens) and has been associated with HLA B51 and Factor V Leiden mutation 2.
Behçet’s disease is classically characterised by the triad of oral ulceration, genital ulceration and ocular lesions. As well as the aforementioned triad of symptoms, Behçet’s is a multi-system disorder and has the potential to involve the cardiovascular system (as in this case), the lungs, the gastrointestinal tract, genitourinary tract, the central nervous system, the skin and joints 3.
Diagnosis of Behçet’s Disease can be difficult and therefore international criteria have been developed for this purpose. The International Criteria for Behçet’s Disease (ICBD) and associated scoring is as follows, with a score of ≥4 being diagnostic for Behçet’s 4:
- Ocular lesions – 2 points
- Genital aphthosis – 2 points
- Oral aphthosis – 2 points
- Skin lesions – 1 point
- Neurological manifestations – 1 point
- Vascular manifestations – 1 point
- Positive pathergy test – 1 point
The patient in this case would have scored 3 points, being positive for oral aphthous ulcers and vascular manifestations. Pathergy refers to an exaggerated reaction to skin injury from minor trauma (such as a needlestick) and this patient was noted to develop thrombophlebitis at a number of peripheral venous cannula sites but had no definite evidence of pathergy.
Treatment and prognosis is variable, with some patients’ disease resolving spontaneously, others requiring long courses of corticosteroid therapy and other’s requiring management with surgery or interventional radiology. If the disease does resolve it also has the potential to recur at a later date 5.
Hughes-Stovin syndrome is a multi-system inflammatory condition characterised by pulmonary/bronchial artery aneurysm in the setting of thrombophlebitis. Radiological findings and histopathological findings can be similar or sometimes indistinguishable from Behçet’s Disease, with the result that the term ‘incomplete Behçet’s Disease’ has been used to describe Hughes-Stovin Syndrome. Some authors postulate that Hughes-Stovin syndrome is in fact Behçet’s Disease 6 and even without the other ICBD criteria that it should be treated as such.
Questions
- Which human leukocyte antigen (HLA) type is associated with development of Behçet’s Disease?
- HLA-B27
- HLA-B47
- HLA-B51
- HLA-DR3
- HLA-DR4
2. What is the most common symptom in patients presenting with Behçet’s Disease
-
- Genital aphthous ulcers
- Ocular manifestations (e.g. uveitis or retinal vasculitis)
- Oral aphthous ulcers
- Joint manifestations (arthralgia or arthritis)
- Vascular manifestations
3. Behçet’s Disease is a vasculitis that can affect large, medium and small vessels. Which of these vasculitides is classically considered a large vessel vasculitis?
-
- Eosinophilic granulomatosis with polyangiitis
- Granulomatosis with polyangiitis
- IgA vasculitis
- Kawasaki disease
- Takayasu’s arteritis
Answers
- C.
HLA-B51 is associated with the development of Behçet’s Disease 2. HLA-B27 is associated with ankylosing spondylitis. HLA-B47 is associated with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. HLA-DR3 is associated with Sjogren’s syndrome, autoimmune hepatitis and type 1 diabetes. HLA-DR4 is associated with type 1 diabetes and rheumatoid arthritis 7.
- C.
Oral aphthous ulcers are the most common symptom in patients presenting with Behçet’s Disease. When creating the International Criteria for Behçet’s Disease (ICBD), 1278 patients with Behçet’s Disease from 27 countries were identified. 98% of these had oral aphthous ulcers, 74% had genital aphthous ulcers, 55% had ocular manifestations, 51% had joint manifestations and 19% had vascular manifestations 4.
- E.
Takayasu’s arteritis is a large vessel vasculitis. Eosinophilic granulomatosis with polyangiitis (previously Churg-Strauss syndrome), granulomatosis with polyangiitis (previously Wegener’s granulomatosis) and IgA vasculitis (previously Henoch-Schönlein purpura) are all small vessel vasculitides. Kawasaki disease is a medium vessel vasculitis.
_______________________
References
- Kontogiannis V, Powell RJ. Behçet’s disease. Postgrad Med J. 2000 Oct; 76(900): 629–637.
- Marshall SE. Behçet’s disease. Best Pract Res Clin Rheumatol. 2004 Jun;18(3):291-311.
- Davatchi F, Chams-Davatchi C et al. Behçet’s disease: epidemiology, clinical manifestations, and diagnosis. Expert Rev Clin Immunol. 2017 Jan;13(1):57-65.
- International Team for the Revision of the International Criteria for Behçet’s Disease (ITR-ICBD).The International Criteria for Behçet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014 Mar;28(3):338-47.
- Cheon JH, Kim WH. An update on the diagnosis, treatment, and prognosis of intestinal Behçet’s disease. Curr Opin Rheumatol. 2015 Jan;27(1):24-31.
- Erkan D, Yazici Y et al. Is Hughes-Stovin syndrome Behçet’s disease? Clin Exp Rheumatol. 2004 Jul-Aug;22(4 Suppl 34):S64-8.
- Kumar V, Robbins SL. Table 5-7. Robbins Basic Pathology (8th edition). 2007. Philadelphia: Elsevier Saunders.


